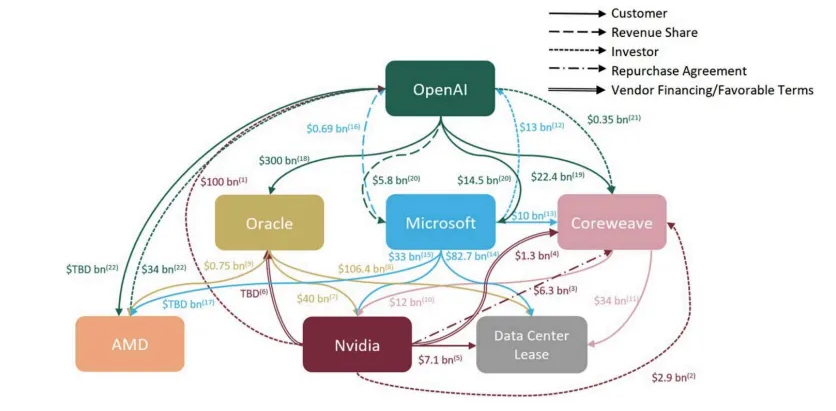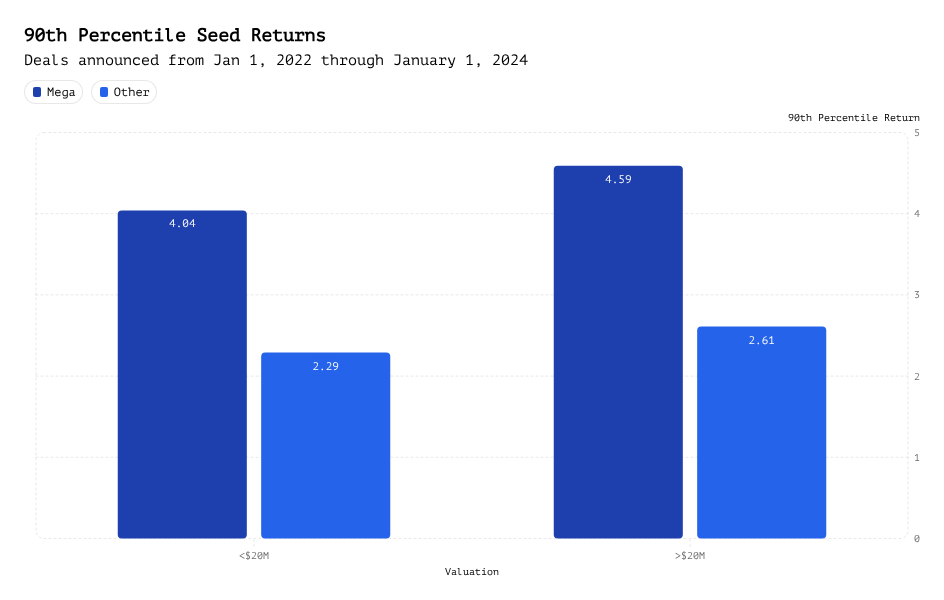
Mega Funds at Seed: Existential Risk or Smart Beta Opportunity?
December 18, 2025
Level recently invested in Enveda Biosciences‘ Series B. We are excited about the company’s potential, and the use of advanced computational methods to find therapeutic solutions to human disease.
Enveda searches for potential medicines by analyzing complex chemical samples derived from nature (plant samples). Without going into too much detail, nature is, intuitively, an excellent source for potentially new drugs. Naturally occurring compounds have been optimized by evolution, over hundreds of millions of years, to have specific bioactivities (which can have efficacy against human disease targets). Despite natural compounds being only a tiny portion of theoretical compounds (10^60), they contribute more than 50% of FDA approved drugs. Enveda believes that a major opportunity exists in analyzing the thousands of (most unknown) metabolites that exist in natural samples.
At Level we attempt to harness complex networks and graphs. Networks exist all around us, and we believe framing machine learning problems as graph learning problems can provide unique value and performance gains (relational inductive bias). Within our own industry (venture capital), we have developed a suite of algorithms that construct networks (from raw data streams) and run algorithms on on top of them. As a team, we continuously think about network dynamics, complexity, non-equilibrium, and power laws.
Enveda recently published a post detailing GRAFF-MS, which utilizes graph neural networks as a core primitive for predicting mass spectra from molecular structure (this is building on their early work, called MS2Prop, which predicts chemical properties from mass spec data). This technique can be thought of as inverse of structural elucidation, where the idea is to augment libraries with spectra predicted from large databases of molecular graphs. There are few (10^4) small molecules with known experimental mass spectra, making augmentation incredibly valuable.
A key challenge in predicting spectra given molecular structure is the nature of the output space, which in the case of mass spec requires distinguishing m/z differences on the order of 10^-6. One of the existing methods for predicting spectra include bond-breaking. Bond-breaking enumerates the 2D structure of all probably product ions, using edge removals of the molecular graph. Among other things, this is a computationally slow process (~5 seconds to predict a single mass spectrum, which would take three months on a 64-core machine for the ~300k spectra in NIST-20!).
We will focus on the graph learning component, but below is a high-level summary of the GRAFF-MS approach:
We will focus on the last item, the GNN, employs the following in its core architecture:
We believe the above graph neural network approach is interesting in several ways:
As these graph neural networks become more sophisticated and supporting libraries like DGL continue to mature for life sciences use cases, we will see incredible progress in computational drug discovery.